Autres noms Tumeur de la glande pinéale, pinéaloblastome
Qu'est-ce que le pinéoblastome ?
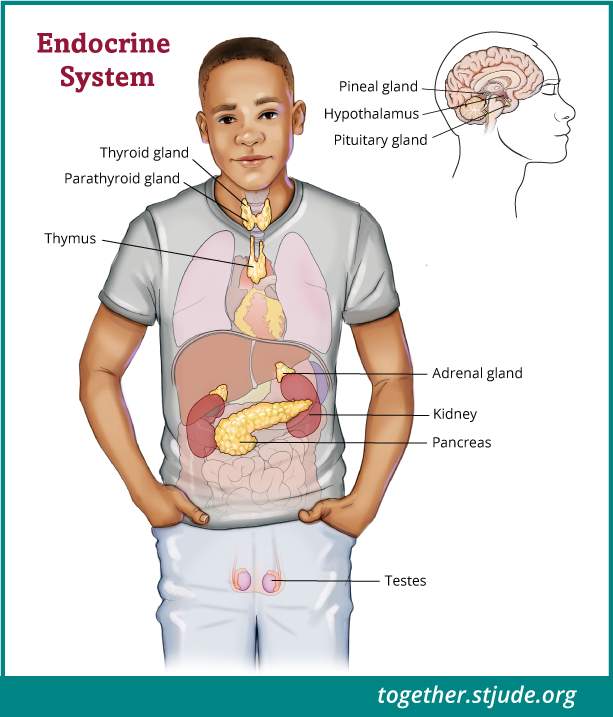
Le pinéoblastome est une tumeur cérébrale rare qui peut survenir chez les enfants et les jeunes adultes. Cette tumeur apparaît dans la glande pinéale, une petite structure située profondément dans le cerveau.
Environ 30 % (3 sur 10) des tumeurs trouvées dans la glande pinéale sont des pinéoblastomes. En raison de leur emplacement, ces tumeurs peuvent être difficiles à traiter. Les tumeurs de pinéoblastome représentent moins de 1 % des tumeurs cérébrales chez les enfants. Le taux de survie à 5 ans pour le pinéoblastome pédiatrique varie de 67 à 85 % aux États-Unis, selon le type.
La glande pinéale libère l'hormone mélatonine, qui contrôle le sommeil. La mélatonine aide également à contrôler la libération des hormones de reproduction, de la glande pituitaire, comme l’hormone lutéinisante (LH) et l’hormone folliculo-stimulante (FSH). Ces hormones sont importantes pour la fertilité et la santé reproductive. Certains traitements du pinéoblastome peuvent affecter la fertilité.
Le pinéoblastome est une tumeur rare qui se développe dans l'épiphyse dans le cerveau.
Symptômes du pinéoblastome
Les signes et symptômes du pinéoblastome dépendent de la taille de la tumeur et si elle s’est propagée dans d'autres parties du cerveau. Souvent, la tumeur qui croit provoque le liquide céphalorachidien une accumulation de liquide dans le cerveau, ce qui exerce une pression. Ceci est connu sous le nom d'hydrocéphalie. La pression accrue peut provoquer certains symptômes qui se manifestent avec des tumeurs pinéales.
Les symptômes du pinéoblastome peuvent inclure :
Les pupilles qui sont plus grandes que la normale ou ont des tailles différentes
Changements dans les mouvements oculaires, en particulier des problèmes pour regarder vers le haut
Maux de tête
Déshydratation
Soif supérieure à la normale
Énurésie qui commence soudainement
Mictions fréquentes (uriner) la nuit
Nausées et vomissements
Changements dans le niveau d'énergie
Fatigue
Problèmes de coordination, d'équilibre ou de mouvement
Faiblesse d'un côté du corps.
Convulsions
Changements de comportement
Taux de croissance lent
L'accumulation de liquide dans le cerveau peut exercer une pression. Cette condition est connue sous le nom d'hydrocéphalie et peut nécessiter un traitement.
Facteurs de risque du pinéoblastome
Souvent, les médecins ne savent pas ce qui cause le pinéoblastome. Mais certains facteurs entraînent un risque accru. Le pinéoblastome peut :
Être plus commun à certains âges qu'à d'autres, selon le type.
Se produire chez des enfants atteints de rétinoblastome bilatéral.
L'équipe de soins de votre enfant peut référer votre famille à un conseiller en génétique si nécessaire.
Diagnostic du pinéoblastome
Un diagnostic de pinéoblastome nécessite des tests et des procédures. Le médecin fera passer un examen physique à votre enfant, posera des questions sur ses antécédents médicaux et demandera des tests tels que :
Biopsie pour vérifier la présence de cellules cancéreuses
Tests d’imagerie
Examen neurologique avec des tests qui vérifient comment le cerveau et la moelle épinière fonctionnent
Ponction lombaire pour rechercher des cellules cancéreuses dans le liquide céphalorachidien (LCR)
Des examens sanguins pour rechercher les marqueurs tumoraux (des substances libérées par certaines tumeurs)
Des tests génétiques pour rechercher des modifications de l'ADN des cellules
Types de pinéoblastome
Il existe différents types de pinéoblastome en fonction des changements génétiques (mutations) qui causent la tumeur. La tumeur de votre enfant nécessite des tests moléculaires pour un diagnostic précis et pour déterminer le meilleur traitement.
Les types de pinéoblastome chez l’enfant incluent :
Pinéoblastome-miRNA1
Pinéoblastome-miRNA2
Pinéoblastome, FOXR2 activé
Pinéoblastome, RB1 activé
Naviguer dans une maladie grave avec des enfants d'âge scolaire
Élever un enfant d'âge scolaire pendant une maladie peut être accablant. Elnora Lee aborde l'éducation de trois filles en âge scolaire tout en s'occupant des soins et du traitement d'une maladie grave.
Ce cancer reste généralement dans le cerveau et la moelle épinière (système nerveux central). Dans 10 à 20 % (1 à 2 sur 10) des cas, le pinéoblastome se propage par le liquide céphalorachidien vers d'autres parties du corps.
Le pinéoblastome est classé par métastase, ou propagation de la maladie :
M0 : La tumeur est à un endroit sans signe de propagation à d'autres parties du corps.
M1 : Les cellules tumorales se sont propagées au liquide céphalo-rachidien (LCR).
M2 : La tumeur s'est propagée à d'autres parties du cerveau.
M3 : La tumeur s'est propagée à la colonne vertébrale.
M4 : La tumeur s'est propagée en dehors du système nerveux central (SNC).
Les tumeurs de la glande pinéale sont également classées en fonction de leur apparence au microscope, et de leur vitesse de croissance et de propagation dans le corps. Plus les cellules tumorales sont anormales à l'observation, plus le grade de la tumeur est élevé. Les pinéoblastomes sont des tumeurs de haut grade (grade 4).
Traitement du pinéoblastome
Le traitement du pinéoblastome dépend du :
Type du pinéoblastome
Âge et état de santé du patient lors du diagnostic
Si le cancer s'est propagé
Si la tumeur peut être retirée par chirurgie
Si le patient est assez âgé pour recevoir une radiothérapie
L'objectif de la chirurgie est de retirer un maximum de la masse tumorale. La chirurgie pour le pinéoblastome peut être difficile en raison de l'emplacement de la tumeur et du risque pour les vaisseaux sanguins voisins. Dans certains cas, il n'est pas possible d'enlever toute la tumeur en une seule intervention. Dans de tels cas, les chirurgiens peuvent décider de :
Faire à la fois une biopsie et une chimiothérapie afin que les chirurgiens puissent ensuite retirer complètement la tumeur.
Certains patients atteints de pinéoblastome peuvent subir une chirurgie pour aider à réduire les effets secondaires de l'hydrocéphalie. Un chirurgien fera une petite ouverture dans le troisième ventricule du cerveau. Cette ouverture aide le liquide céphalo-rachidien à circuler plus librement pour éviter qu'il ne s'accumule et ne cause des dommages. Dans d'autres cas, les médecins peuvent placer un shunt, un petit tube qui draine le liquide céphalo-rachidien du cerveau. Le shunt peut être temporaire ou permanent.
Radiothérapie
La radiothérapie est souvent donnée après une chirurgie pour le pinéoblastome. L'énergie provenant des rayonnements peut endommager ou détruire les cellules cancéreuses à croissance rapide. Les médecins peuvent également utiliser la radiothérapie pour réduire la tumeur. La dose et la quantité de radiothérapie dépendent de :
L'âge de votre enfant
du type de tumeur
Si la maladie s’est propagée
Les rayonnements ne sont pas utilisés chez les jeunes enfants, car ils augmentent le risque d'effets secondaires à long terme. Chez les patients de plus de 3 ans, la radiothérapie est généralement administrée à l'ensemble du système nerveux (irradiation craniospinale). Une dose plus élevée (renfort) est administrée sur le site de la tumeur primaire.
En savoir plus sur la sécurité radiologique.
Chimiothérapie
La chimiothérapie est souvent utilisée en association avec la radiothérapie et la chirurgie :
Avant la chirurgie, les médecins utilisent parfois la chimiothérapie pour réduire la tumeur afin qu'elle soit plus facile à enlever.
Après la chirurgie, la chimiothérapie peut détruire les cellules cancéreuses qui restent.
Chez les très jeunes enfants, la chimiothérapie peut être utilisée pour ralentir la croissance du cancer jusqu'à ce que l'enfant soit suffisamment âgé pour recevoir des rayonnements en toute sécurité.
Plusieurs médicaments de chimiothérapie peuvent être utilisés en même temps pour traiter le pinéoblastome. Les traitements courants sont la vincristine, le cyclophosphamide et le cisplatine. L'équipe de soins de votre enfant peut utiliser d'autres types de chimiothérapie.
Le pronostic du pinéoblastome
L’ pronostic dépend de nombreux facteurs. Le taux de survie à 5 ans pour le pinéoblastome pédiatrique varie de 67 à 85 % aux États-Unis, en fonction du type, de l'âge de votre enfant et des traitements reçus.
L’équipe soignante de votre enfant est la meilleure source d’information sur le cas de votre enfant.
Les facteurs qui affectent les chances de guérison sont les suivants :
Type de pinéoblastome
L’âge au moment du diagnostic
Si le cancer s’est développé.
Si la tumeur peut être retirée par chirurgie
Si le patient est assez âgé pour recevoir une radiothérapie
Si la tumeur a été nouvellement diagnostiquée ou est revenue (récidivée)
L'ablation réussie de la tumeur et le traitement par radiothérapie sont liés à de meilleurs résultats pour les enfants atteints de pinéoblastome.
Soutien pour les patients atteints de pinéoblastome
Faire face à un diagnostic de cancer et à un traitement peut être stressant pour vous et votre famille. Vous pourriez vouloir parler avec un travailleur social, un psychologue ou un autre spécialiste de la santé mentale.
Après le traitement, le médecin de votre enfant peut utiliser des tests d'imagerie et des examens pour surveiller la récidive. Votre enfant pourrait également avoir besoin d'un traitement pour des problèmes neurologiques, cognitifs et endocriniens.
Certains traitements peuvent entraîner des effets tardifs. Il s’agit de problèmes de santé qui surviennent des mois, voire des années, après la fin du traitement. Les patients qui reçoivent une radiothérapie pour la glande pinéale sont à risque de problèmes endocriniens à long terme, tels qu'une faible fonction pituitaire. Les glandes endocrines produisent des hormones qui contrôlent les fonctions corporelles. Cela peut entraîner des problèmes tels que :
La surveillance continue des niveaux hormonaux est importante. Les patients peuvent avoir besoin de médicaments, y compris des traitements hormonaux substitutifs, pour remplacer les hormones naturelles manquantes lorsque le corps ne produit pas ce dont il a besoin.
Les patients qui reçoivent une radiothérapie de la glande pinéale risquent des changements endocriniens à long terme.
Après avoir terminé le traitement, il est important de :
Bénéficier de bilans de santé et de dépistages réguliers par un médecin traitant.
Maintenir des habitudes saines, y compris l'activité physique et une alimentation saine.
Avoir un plan de soins de survie à partager avec les prestataires de santé. Ce plan doit inclure :
Orientation sur les dépistages de santé
Facteurs de risque de la maladie
Comment améliorer sa santé
Les survivants d’un cancer de l’enfant devraient bénéficier d’un suivi médical à long terme.
Questions à poser à votre équipe soignante
Quelles sont les options de traitement de mon enfant ?
Quels sont les effets secondaires possibles de chaque traitement ?
Mon enfant devra-t-il être hospitalisé pour son traitement ?
Où le traitement est-il disponible ? Est-ce proche de chez nous ou bien devrons-nous nous déplacer ?
Quelles ressources pouvons-nous utiliser pour aider à faire face à cette maladie ?
Points clés sur le pinéoblastome
Le pinéoblastome est une tumeur cérébrale rare de la glande pinéale, qui est située profondément dans le cerveau.
Ces tumeurs peuvent être difficiles à traiter en raison de leur emplacement.
Le diagnostic peut nécessiter un examen physique, une biopsie, des tests d'imagerie, un examen neurologique, une ponction lombaire, des analyses de sang et des tests génétiques.
Le traitement dépend de la tumeur, mais peut inclure la chirurgie, la radiothérapie et la chimiothérapie.
Le taux de survie pour les cas de pinéoblastome nouvellement diagnostiqués est d'environ 67 à 85 % aux États-Unis. Cela dépend du type de maladie, de l'âge et de la santé de votre enfant, ainsi que du type de traitement qu'il reçoit. Discutez du pronostic de votre enfant avec son prestataire de soins.
Les tumeurs cérébrales et de la moelle épinière peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). En savoir plus sur les symptômes, le diagnostic et le traitement des tumeurs cérébrales chez les enfants et les adolescents.
Les traitements qui guérissent le cancer peuvent également avoir certains effets secondaires tardifs et à long terme. En savoir plus sur les traitements qui sont liés à certains effets tardifs.
Le médulloblastome est la tumeur cérébrale maligne la plus fréquente chez l'enfant. C'est une tumeur à croissance rapide qui apparaît dans le cervelet à l'arrière du cerveau.