Qu’est-ce qu’un neuroblastome ?
Le neuroblastome est un cancer qui se développe à partir de cellules du système nerveux. Il se forme lorsque les cellules saines du système nerveux ne se développent pas comme elles le devraient. Au lieu de cela, des cellules cancéreuses appelées « neuroblastes » restent bloquées à un stade précoce de développement. Elles ne deviennent pas des cellules saines et normales. Les cellules cancéreuses commencent à se développer, ce qui provoque la formation d’une tumeur.
Les patients atteints de neuroblastome peuvent n’avoir qu’une seule tumeur. Ou bien les cellules de neuroblastome peuvent se propager à d’autres parties du corps. Le neuroblastome affecte généralement les enfants de moins de 5 ans. C’est la tumeur solide la plus fréquemment observée en dehors du cerveau chez les enfants.
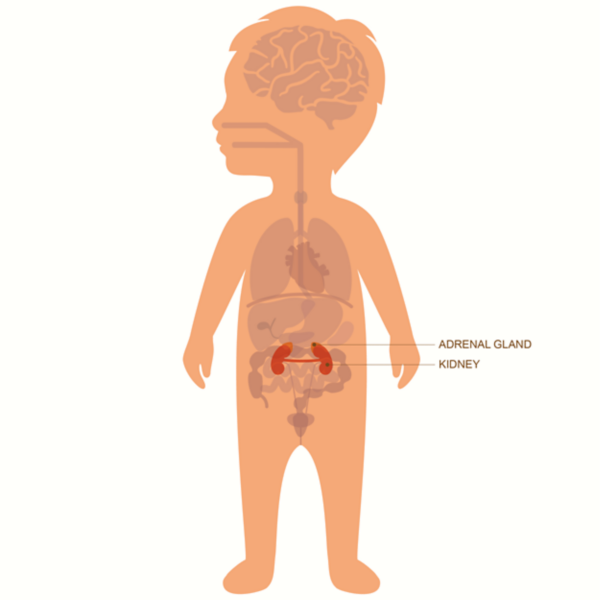
Le neuroblastome commence souvent dans l’abdomen, soit dans une glande surrénale, soit dans d’autres cellules nerveuses. Il peut également se former dans le cou, la poitrine ou le bassin.
Les symptômes dépendent de l’emplacement de la tumeur (ou des tumeurs si le cancer s’est propagé). Les symptômes peuvent inclure une grosseur, de la douleur, une perte d’appétit, de la fatigue et de l’irritabilité.
- Certains bébés atteints de neuroblastome n’ont pas besoin de traitement. Le médecin suivra de près l’évolution de la tumeur, et cette dernière pourra parfois disparaître d’elle-même.
- Certains enfants atteints de neuroblastome n’ont besoin que d’une intervention chirurgicale.
- Cependant, environ la moitié des patients atteints de neuroblastome présentent une maladie à risque élevé. Ils ont besoin de chimiothérapie, d’une intervention chirurgicale, de radiothérapie et d’immunothérapie.
Chaque année, environ 700 enfants reçoivent un diagnostic de neuroblastome aux États-Unis.
Symptômes du neuroblastome
Les signes et symptômes dépendent de l’emplacement du cancer. Ils peuvent inclure :
- Grosseur ou masse au niveau du cou, du thorax ou de l’abdomen
- Gonflement des yeux ou cernes autour des yeux
- Maux d’estomac
- Irritabilité
- Diminution de l’appétit
- Constipation
- Sensation de fatigue
- Faiblesse dans les jambes
Les autres symptômes peuvent inclure :
- Diarrhée
- Changement du mouvement des yeux
- Hypertension artérielle
- Maux de tête
- Toux
- Difficulté à respirer
- Fièvre
- Ecchymoses
- Syndrome de Horner
Le neuroblastome s’est souvent propagé à d’autres parties du corps avant d’être diagnostiqué.
Qu’est-ce que le syndrome de Horner ?
Certains patients atteints de neuroblastome développent le syndrome de Horner, ou des lésions nerveuses autour de l’œil. Les symptômes peuvent inclure :
- Une paupière tombante
- Pupille de petite taille
- Perte de la capacité de transpirer d’un côté du visage
En savoir plus sur le syndrome de Horner
Facteurs de risque et causes du neuroblastome
Le neuroblastome est le plus fréquent chez les jeunes enfants. Il survient un peu plus souvent chez les garçons que chez les filles.
Un petit nombre de patients (1 à 2 %) ont un neuroblastome héréditaire. Celui-ci peut se transmettre de génération en génération. Il est souvent causé par un changement dans les gènes ALK ou PHOX2B. D’autres mutations génétiques ont toutefois également été liées au neuroblastome héréditaire.
En savoir plus sur le neuroblastome héréditaire
Diagnostic du neuroblastome
Les médecins utilisent plusieurs types de tests lors du diagnostic du neuroblastome. Les tests peuvent inclure :
- Un examen physique et les antécédents médicaux
- Des analyses de sang pour vérifier la numération globulaire, la fonction rénale et la fonction hépatique
- Des analyses d’urine pour rechercher l’acide vanillylmandélique (VMA) et l’acide homovanillique (HVA). Le VMA et l’HVA sont produits par la décomposition des catécholamines, qui sont des hormones produites par les neuroblastes. Les enfants atteints de neuroblastome ont souvent un taux élevé de VMA et d’HVA. Ces substances peuvent permettre pour surveiller la réaction au traitement.
- Un examen neurologique. pour mesurer différents aspects de la fonction cérébrale et nerveuse, notamment la mémoire, la vision, l’audition, la force musculaire, l’équilibre, la coordination et les réflexes
- Tests d’imagerie pour connaître la taille et l’emplacement de la ou des tumeurs
Les médecins examineront la tumeur à la recherche de caractéristiques importantes pour diagnostiquer et traiter le neuroblastome. Certaines tumeurs sont agressives et nécessitent une thérapie intense. Les médecins peuvent prévoir la réponse d’une tumeur au traitement selon l’aspect des cellules et la présence de certaines mutations génétiques dans la tumeur.
La stadification du neuroblastome est effectuée à l’aide du système de stadification du groupe international de risque du neuroblastome (International Neuroblastoma Risk Group Staging System, INRGSS). La stadification est basée sur ces facteurs :
- Emplacement de la tumeur
- Effet sur les organes voisins
- Propagation de la maladie
Groupes de risque du neuroblastome
Les médecins utilisent également des groupes de risque pour classer les cas de neuroblastome et planifier les traitements. Il existe 3 groupes de risque :
- Risque faible
- Risque intermédiaire
- Risque élevé
Le neuroblastome à risque élevé signifie que le cancer est difficile à retirer et qu’il a plus de chance de récidiver. Les enfants présentant un neuroblastome à risque élevé ont besoin d’une thérapie intense.
Les groupes de risque sont basés sur des facteurs qui incluent les suivants :
- Âge du patient
- Stade de la maladie
- Caractéristiques de la tumeur
Traitement du neuroblastome
Le traitement dépend du groupe de risque assigné. Les options de traitement peuvent inclure :
Les patients très jeunes à faible risque peuvent être mis en observation plutôt que de recevoir un traitement actif. Parfois, le neuroblastome disparaît de lui-même (régression), mais cela est rare. Les patients sont surveillés de près pour observer la progression de la tumeur..
Les patients atteints de neuroblastome peuvent se voir proposer un traitement dans le cadre d’un essai clinique. Les traitements du neuroblastome sont planifiés en se basant sur les groupes de risque :
Pronostic du neuroblastome
La survie au neuroblastome dépend de plusieurs facteurs :
- Âge de l’enfant au moment du diagnostic
- Groupe de risque
- La génétique de la tumeur
- Si le cancer s’est propagé
- Réponse de la tumeur au traitement
- Si le cancer est récidivant (est réapparu)
Le médecin de votre enfant est votre meilleure source d’information concernant le cas spécifique de votre enfant.
Accompagnement des patients atteints de neuroblastome
Surveillance pour détecter la survenue d’une rechute
Les patients ont besoin de soins de suivi afin de détecter la survenue éventuelle d’une récidive après la fin du traitement. L’équipe médicale suggérera des tests spécifiques et leur calendrier.
Chez les patients qui n’ont pas une maladie à risque élevé, le risque de rechute est de 5 à 15 %. Chez les patients à risque élevé, le risque de rechute est d’environ 50 %. La rechute survient le plus souvent au cours des 2 premières années après le traitement. La rechute survient rarement en l’absence de signe de cancer 5 ans après la fin du traitement.
La santé après un cancer
Les survivants traités par chimiothérapie ou radiothérapie doivent faire l’objet d’une surveillance pour détecter des effets à long terme et tardifs de la thérapie. Les problèmes dus au traitement pourraient inclure une perte auditive, des problèmes cardiaques, des problèmes de thyroïde, un ralentissement de la croissance, une diminution de la densité osseuse et des lésions rénales.
Environ 25 % des survivants souffrent de maladies chroniques graves 25 ans après le diagnostic, selon l’Étude sur les survivants d’un cancer de l’enfant.
Il est important qu’un prestataire de soins de santé traitant effectue des bilans de santé réguliers pour surveiller les problèmes de santé susceptibles de se développer plusieurs années après la thérapie.
L’équipe de soins de votre enfant doit vous fournir un plan de soins de survie après la fin du traitement. Ce rapport comprendra les tests nécessaires et des conseils pour adopter un mode de vie sain.
—
Révision : Novembre 2023